
Практичне заняття 6. Генні хвороби
1. Загальна характеристика і класифікація генних хвороб.
2. Механізми виникнення моногенних хвороб.
3. Особливості діагностики та лікування генних хвороб.
4. Медико-генетичне консультування при генних хворобах.
Література: Медична
біологія / За ред. В.П.Пішака та Ю.І.Бажори. – Вид. 3-тє. – Вінниця: Нова
книга, 2017.С. 216-229
1. Загальна характеристика і класифікація генних хвороб.
Генні захворювання – це спадкові аномалії, викликані наявністю дефектного гена, який успадковуються, як менделівські ознаки.
Для вивчення генного захворювання необхідно знайти мутантний алель з патологічною дією, визначити первинний ефект мутантного гена (початкова ланка патогенеза), розшифрувати взаємодію генів між собою та з середовищем (патогенез захворювання в цілому). Саме на цих даних базується профілактика, діагностика, лікування генних захворювань.
Більшість відомих генних хвороб викликані мутаціями в структурних генах. Існують чіткі докази впливу певних змін послідовності нуклеотидів на виникнення деяких хвороб. Можливість впливу мутацій генів-регуляторів на формування генних хвороб також існує, але ці дефекти вивчені менше.
Класифікація генних захворювань базується на дії дефектного гена. Більшість моногенних хвороб - це порушення обміну речовин.
Класифікація молекулярних порушень обміну речовин (з підручника за ред. В.П.Пішака та Ю.І.Бажори).
1. Порушення метаболізму амінокислот:
1.1. Фенілаланіну (фенілкетонурія);
1.2. Тирозину (тирозинемія, алькаптонурія);
1.3. Метіоніну (гомоцистинурія);
1.4. Цистину (цистинурія);
1.5. Триптофану (хвороба Хартнупа, триптофанемія та ін.);
1.6. Лейцину (хвороба кленового сиропу);
1.7. Гістидину (гістидинурія, гістидинемія)
та інших амінокислот.
2. Порушення метаболізму вуглеводів:
2.1. Галактози (галактоземія);
2.2. Фруктози (фруктоземія);
2.3. Глікогену (глікогенози);
2.4. Дисахаридозні ентеропатії (синдром мальабсорбції вуглеводів).
3. Спадкові хвороби обміну сполучної тканини:
3.1. Мукополісахаридози;
3.2. Хвороба Марфана.
4. Спадкові хвороби обміну ліпідів:
4.1. Гіперліпопротеїнемії;
4.2. Сфінголіпідози (хвороба Німанна - Піка);
4.3. Гангліозидози (хвороба Тея - Сакса).
5. Спадкові хвороби порфіринового обміну (порфірії).
6. Ензимопатії жовчно-пігментного обміну (хвороба Жильбера).
7. Ензимопатії панкрео-інсулярного гормоносинтезу:
7.1. Муковісцидоз;
7.2. Уроджена відсутність ензимів підшлункової залози;
7.3. Хвороба Вільсона - Коновалова;
7.4. Целіакія.
8. Ензимопатії біосинтезу гормонів.
Інший варіант класифікації спадкових вад метаболізму в людини.
1. Порушення обміну амінокислот (фенілкетонурія, альбінізм, аргінінемія, гістидінемія та ін.).
2. Порушення обміну вуглеводів (галактоземія, непереносимість дисахаридів).
3. Порушення обміну ліпідів (хвороби Тея-Сакса, Гоше, Німана-Піка).
4. Порушення обміну стероїдів (адрено-генітальний синдром, чоловічий псевдогермафродитизм).
5. Порушення обміну пуринів і піримідинів (ксантинурія).
6. Порушення обміну речовин сполучної тканини, кісток і м'язів (мукополісахаридоз).
7. Порушення обміну гему і порфірину (порфірія).
8. Порушення обміну речовин еритроцитів (гемоглобінопатії, гемолітичні анемії).
9. Порушення обміну металів (гепатолентикулярна дегенерація).
10. Порушення транспорту речовин (лізінурія).
11. Порушення ферментів і білків плазми (коагулопатії, недостатність ферментів).
Загальна частота генних хвороб у популяціях становить 1-2%. Частота окремих хвороб може значно коливатись. умовно частоту генної хвороби можна вважати високою, якщо зустрічається одна хвора дитина на 10 тис. або менше новонароджених, середньою - на 10-40 тис., низькою - більше, ніж на 40 тис. Зрозуміло, що частота генних хвороб у більш старших вікових групах буде змінюватись з урахуванням частоти летальних випадків.
Для конкретної генної хвороби частота може значно змінюватись в залежності від того, яка популяція людини розглядається. Так, частота фенілкетонурії в більшості європейських країн та США становить 1:10-15 тис., але у Швеції та Фінляндії вона значно менша (1:43 тис. та 1:71 тис. відповідно), а в Японії - це взагалі рідка хвороба (1:210 тис.). Частота муковісцидозу у європейців становить 1:2500-5000, а у чорношкірих африканців - 1:77 тис.
2. Механізми виникнення моногенних хвороб.
Механізм виникнення генного захворювання залежить від того, яку функцію виконує в організмі білок, що кодується дефектним геном.
Прикладом дефекту структурного білку може бути синдром Елерса-Данлоса, при якому змінена молекулярна структура колагену (волокнистого білку, що забезпечує механічну міцність міжклітинної речовини). Прояви: підвищена еластичність шкіри, збільшена рухомість суглобів, відшарування сітківки, підвивих кришталика.
До порушень синтезу гемоглобіну відносяться, зокрема, серпоподібноклітинна анемія та таласемія.
Таласемія – це спадкове захворювання крові, яке характеризується продукцією аномального гемоглобіну. Причиною є дефекти генів, які призводять до порушення синтезу одного із чотирьох ланцюгів, які складають молекулу гемоглобіну. Цими дефектами можуть бути як делеції структурних генів, так і синтез аномальної матричної РНК, неефективна транскрипція або мутації регуляторних генів. В результаті спостерігається значне зниження або відсутність синтезу одного із поліпептидних ланцюгів, які складають молекулу гемоглобіну, при збільшенні синтезу решти трьох ланцюгів. Відбувається деформація молекули гемоглобіну, що призводить до деформації еритроцитів, порушення їх функції, та як кінцевий наслідок — їх руйнування та загибель. Наслідком підвищеного гемолізу еритроцитів є анемія. Існує кілька форм таласемії (в залежності від того, синтез якого ланцюга гемоглобіну порушений та від ступеня порушення). Клінічні прояви таласемії залежать від форми захворювання. При важких гомозиготних формах таласемії спостерігається водянка плоду та народження мертвої дитини. Гетерозиготні за геном таласемії особи мають більшу стійкість до захворювання тропічною малярією.
Найбільшу долю генних захворювань складають ензимопатії (дефекти ферментів). Розглянемо загальний механізм їх виникнення на прикладі галактоземії. Галактоза – це складова частина лактози (молочного цукру). В організмі вона перетворюється на глюкозо-6-фосфат. При галактоземії внаслідок дефекту одного з ферментів процес зупиняється на стадії галактозо-6-фосфату. В результаті токсичної дії цієї речовини розвивається цироз печінки, уражуються нирки. З галактози утворюється дульцитол, накопичення якого в кришталику приводить до ранньої катаракти. У дітей внаслідок непереносимості материнського молока спостерігається диспепсія, жовтяниця, затримка фізичного та психічного розвитку. Без необхідного лікування діти вмирають у перші місяці життя. У тих, що вижили, розвиваються мікроцефалія, анемія, судоми. Своєчасна діагностика та спеціальне годування запобігають формуванню більшості симптомів хвороби.
Ціла низка генних хвороб пов’язана з порушеннями обміну фенілаланіну та тирозину (рис. 1)
Фенілкетонурія (фенілпіровиноградна олігофренія, хвороба Фелінга).
Клінічні дані. Проявляється з 3-6 місяців посвітлінням волосся, райдужки, специфічним запахом сечі та поту, часто – повнотою, ексудативним діатезом. Припинення або різке уповільнення моторного та психічного розвитку, синдром дитячих згинальних судом (найчастіше – серії кивків). Гіпертонія окремих груп м’язів, що спричиняє формування специфічної пози: (підігнуті ноги та зігнуті руки). Можливі гіперкінези, тремор рук, атаксія, інколи парези за центральним типом. Без спеціального лікування розумова відсталість досягає ступеня ідіотії або глибокої імбецильності. Після 3-4 років стан стабілізується. При лікуванні, початому з перших місяців життя, і доведеному до 5-8 років, недоумство не розвивається.
Успадковування. Аутосомно-рецесивне.
Поширення. Популяційна частота 1:10000, серед осіб з олігофренією – 1:100.
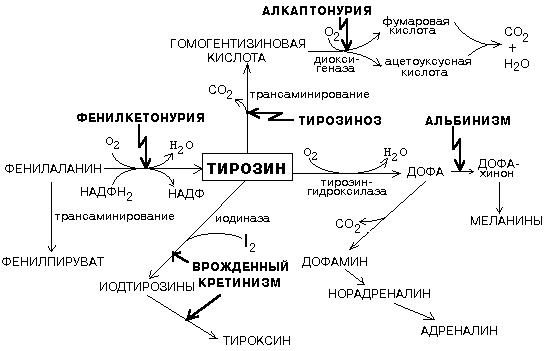
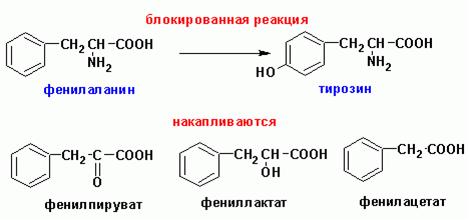
Рис. 1. Порушення обміну амінокислот фенілаланіну та тирозину.
Тирозиноз. Проявляється у перші тижні та місяці життя блювотою, відставанням у фізичному розвитку, збільшенням розмірів печінки. Внаслідок розвитку цирозу з’являється жовтяниця, асцит, крововливи у шкіру, але темпи розвитку ураження печінки можуть варіювати. У зв’язку з ураженням нирок розвивається глюко-аміно-фосфат-діабет, який стає причиною рахітоподібного захворювання кісткової системи (остеопороз та остеомаляція, викривлення довгих трубчастих кісток. У багатьох хворих – затримка розумового розвитку. Протікання хвороби несприятливе, без лікування багато хворих гине у віці до 10 років від печінкової недостатності.
Алкаптонурія. Ознака недостатності ферменту спостерігається відразу після народження: сеча дитини забарвлює пелюшки в чорний колір, тому що гомогентизинова кислота на повітрі окислюється, а потім полімеризується в меланіноподібне сполучення. Екскреція гомогентизинової кислоти залежить від вмісту фенілаланіну і тирозину в їжі.
В дитячому віці алкаптонурія – єдиний прояв дефекту, що клінічно не виявляється. Довготривале накопичення гомогентизинової кислоти призводить до охронозу – пігментації хрящів і сполучної тканини (вушні раковини, склери, шкіри носа, рук і шиї), артропатії. Дегенеративні зміни в пігментованому хрящі обумовлюють розвиток артриту у 50% хворих алкаптонурією у віці 30-40 років.
Альбінізм, тип 1 (повний альбінізм, очно-шкірний тирозиназонегативний альбінізм).
Клінічні дані. Депігментація шкіри, волосся, очей. Їх колір однаковий для всіх рас, з віком не змінюється. Шкіра рожево-червона, зовсім не загоряє. Волосся має жовтуватий відтінок. Хворі схильні до раку шкіри.
Райдужка сіро-блакитна, може бути рожевуватою. Зіничний рефлекс червоний. Ністагм, світлобоязнь, значне зниження гостроти зору, з віком зір не поліпшується. Відсутність бінокулярного зору та макулярного рефлексу.
Успадковування. Аутосомно-рецесивне.
Поширення. 1:39000.
2. Альбінізм, тип 2 (очно-шкірний тирозиназопозитивний альбінізм).
Клінічні дані. Зменшення пігментації залежить від раси, з віком пігмент може накопичуватись, відповідно - колір очей може змінюватись від блакитного до карого, волосся - від білого до жовтувато-коричневого. У 60% хворих – невуси, пігментні плями.
Ністагм і світлобоязнь набагато менші, ніж при альбінізмі першого типу. Але у всіх хворих спостерігається порушення бінокулярного зору, у 90% - відсутність або сильне зниження макулярного рефлексу, косоокість. 20% хворих мають високу міопію. Гострота зору знижена, але з віком підвищується.
Успадковування. Аутосомно-рецесивне.
Серед ензимопатій виділяють лізосомні хвороби нагромадження – це спадкові захворювання, обумовлені порушенням активності ферментів лізосом, що спричиняє накопичення певної групи макромолекул (ліпідів, мукополісахаридів, глікопротеїнів та ін.) в різних органах і тканинах організму. Ці хвороби в переважній більшості аутосомно-рецесивні, зустрічаються рідко, мають прогресуюче протікання, методи лікування для них тільки розробляються.
Розглянемо особливості хвороб нагромадження на прикладі внутрішньоклітинних ліпідозів. Ці хвороби обумовлені дефектом лізосомних ферментів, які розщеплюють жироподібні речовини. В результаті ці речовини накопичуються в лізосомах, що веде до загибелі клітин. Особливо уражується нервова система, виникають як соматичні неврологічні порушення, так і психічні відхилення. Виникають порушення зору, обумовлені патологічними змінами сітківки. Часто спостерігається рання загибель дітей.
Окремі захворювання цієї групи розрізняються за тим, який саме ліпід накопичується.
Хвороба Німана-Піка – відкладається сфінгомієлін. Виділяють три форми цього захворювання:
1. Інфантильна (спленомегалічна) – уражуються головний мозок, збільшуються печінка та селезінка. спостерігається м'язова ригідність, відставання в психічному розвитку, на очному дні формується вишнево-червона пляма. діти гинуть в перші 2-3 роки життя.
2. Підгостра форма – фермент, що розщеплює сфінгомієлін, є, але в зниженій концентрації. Симптоми подібні до першої форми, але менш виражені.
3. Хронічна (вісцеральна) – накопичення сфінгомієліна переважно у внутрішніх органах, що приводить до порушення їх функції.
Успадковування. Аутосомно-рецесивне, хвороба генетично гетерогенна.
Хвороба Гоше. Подібна до хвороби Німана-Піка, але відкладається глюкоцереброзид. Також може проявлятися в трьох формах: гострій, підгострій, хронічній.
Клінічні дані. М'язова ригідність, розумова відсталість, порушення зору, остеопороз, часті переломи кісток, збільшення печінки та селезінки. При гострій формі діти гинуть у перші роки життя, підгостра характеризується більш доброякісним протіканням, хронічна – ураженням внутрішніх органів без патології головного мозку.
Успадковування. Захворювання гетерогенне, описані як аутосомно-рецесивні, так і аутсомно-домінантні варіанти.
Хвороба Тея-Сакса. Характеризується накопиченням гангліозидів. Існує декілька форм цієї хвороби, різних за часом початку та проявами. Загальні симптоми: психічна деградація, втрата зору, судороги, м'язова ригідність.
1. Природжена форма – хвороба Нормана-Вуда.
2. Інфантильна - амавротична ідіотія Тея-Сакса. Це найчастіше захворювання з групи гангліозидозів.
Клінічні дані. Розвивається на другому півріччі першого року життя. У дитини починає знижуватись зір (до сліпоти), психіка збіднюється (до ідіотії), прогресує акінетико-ригідний синдром із судорогами. Підвищена рефлекторна збудливість: доторк або навіть звуки викликають різку рухову реакцію. На очному дні утворюється вишнево-червона пляма з білим вінчиком. Діти гинуть на другому році життя.
Успадковування. Аутосомно-рецесивне. У гетерозигот знаходять вакуолізовані лімфоцити (зі збільшеними лізосомами, наповненими ліпідами).
Поширення. Популяційна частота 1:500000, серед євреїв зі Східної Європи - 1:5000.
3. Пізня інфантильна - хвороба Більшовського.
4. Юнацька - хвороба Шпільмейєра-Фогта.
5. Пізня - хвороба Куфса.
При цих формах інтелект страждає не так сильно, спостерігаються імбецильність або дебільність. Ліпіди відкладаються не тільки в нервовій системі, а й у внутрішніх органах, в першу чергу - печінці та селезінці. Пігментна дегенерація сітківки найбільш виражена при хворобі Шпільмейєра-Фогта.
Існує також велика група генних хвороб, для яких первинний дефектний продукт ще не встановлений. Прикладом може бути муковісцидоз, одне з найпоширеніших генних захворювань. Для нього характерне порушення діяльності всіх екзокринних залоз, що проявляється в підвищеній густоті і в'язкості їх секретів. В результаті виникають численні вторинні порушення в легенях, підшлунковій залозі, кишечнику. На першому році життя у 85-95% хворих відзначаються легеневі проявлення: хронічні бронхіти, бронхопневмонії. Виникають розлади травлення, цироз печінки. Збільшується вміст електролітів у поті.
3. Особливості діагностики та лікування генних хвороб
У більшості випадків природжені порушення обміну речовин з клінічними наслідками проявляються (або можуть бути виявленими) в період новонароджуваності. Такі немовлята відразу після народження звичайно виглядають здоровими, однак ознаки патології, такі як летаргія, утруднення при годівлі, судоми, блювота та ін., можуть проявитися в них уже через кілька годин. Деякі порушення метаболізму можуть залишитися нерозпізнаними в період новонароджуваності і діагноз може бути поставлений тільки через кілька місяців і навіть років. Ранні клінічні прояви звичайно неспецифічні і можуть бути віднесені до перинатальної патології.
Природжене порушення обміну речовин має розглядатися як можливий стан у будь-якої дитини з одним із зазначених клінічних проявів:
- невизначене відставання розумового, рухового розвитку, судоми;
- незвичайний запах, зокрема, під час гострого захворювання;
- інтермітуючі епізоди необгрунтованої блювоти, ацидозу, порушень психіки, кома;
- ниркова колька, гепатомегалія.
Специфічна діагностика спадкових порушень обміну речовин зі встановленим первинним дефектним продуктом може здійснюватись біохімічними методами.
Важливе значення має скринінгове дослідження на наявність достатньо поширених у популяції генних хвороб, що дозволяє рано розпочати лікування і запобігти формуванню частини симптомів.
Масовий (універсальний) скринінг передбачає обстеження усіх осіб з певної категорії (наприклад, усіх новонароджених). Селективний скринінг проводиться в групах ризику (наприклад, скринінг членів сім'ї у разі виявлення спадкового захворювання).
Наприклад, скринінг на фенілкетонурію може проводитись з використанням проби Фелінга (це якісна реакція на фенілпіровиноградну кислоту). Фенілпіровиноградна кислота утворює з іонами трьохвалентного феруму комплексну сполуку, забарвлену у синьо-зелений колір. Пробу Фелінга можна проводити на фільтрувальному папері. Смужку фільтрувального паперу змочують сечею дитини, висушують на повітрі і наносять краплю 10% розчину FeCl3. Позитивна проба дає синьо-зелене забарвлення. Аналогічну пробу можна проводити на сухій або мокрій дитячій пелюшці.
Деякі біохімічні порушення у новонароджених можна виявляти за допомогою мікробіологічного інгібіторного тесту Гатрі. В ньому використовують бактеріальні культури, які вирощують на живильному середовищі, що містить антиметаболіт певної амінокислоти або вуглеводу (фенілаланіну, лейцину, гістидіну, галактози, фруктози або ін.). Краплину крові дитини переносять на фільтрувальний папір і розміщують його на агаровій культурі. При наявності в крові великої кількості досліджуваної речовини спостерігається швидкий ріст бактеріальної культури.
Велике значення в діагностиці генних хвороб мають молекулярно-генетичні методи, зокрема ПЦР.
Лікування. Тривалий час спадкові захворювання вважались невиліковними. Діагноз спадкової хвороби сприймався як вирок приреченому хворому. Причиною цього були, по-перше, недостатні знання про механізми розвитку цих хвороб (вважалось, що наявність дефекту спадковості обов'язково приведе до формування захворювання); і по-друге, те, що діагностика спадкових аномалій була можлива тільки в той період, коли вони вже проявляються клінічно. Завдяки значним успіхам генетики спадкових захворювань і клінічної медицини тепер можна стверджувати: велика кількість спадкових захворювань успішно лікуються.
Загальні підходи до лікування спадкових хвороб спільні з іншими хворобами. Розрізняють:
1. Симптоматичне лікування – заходи, спрямовані на ліквідацію або полегшення окремих симптомів хвороби.
Симптоматичне лікування є неспецифічним по відношенню до конкретної хвороби, воно не впливає на причину її виникнення і тому не може бути головним. Але фактично симптоматичне лікування є необхідним елементом правильної терапії. Симптоматичне лікування стає тим більш ефективним, чим більше воно враховує особливості розвитку хвороби.
2. Патогенетичне лікування - цілеспрямоване втручання в патогенез захворювання, його розвиток і протікання.
Патогенетичне лікування є найважливішим на сучасному етапі розвитку медичної генетики.
3. Етіологічне лікування - усунення першопричини захворювання, яке приводить до найбільш радикального виліковування.
Цей найефективніший метод для спадкових захворювань поки ще не може бути застосований, оскільки питання про втручання в спадковий апарат людини зараз знаходиться на стадії лабораторних досліджень.
Методи лікування
1. Замісна терапія.
Суть цього методу – введення в організм відсутніх або наявних у недостатній кількості біохімічних субстратів. Класичним прикладом замісної терапії є лікування цукрового діабету. Застосування інсуліну дозволило різко зменшити не тільки смертність, але й інвалідизацію хворих. На прикладі інсулінотерапії можна побачити як переваги, так і недоліки цього методу. До числа останніх відноситься необхідність постійного введення в організм дефіцитної речовини. Дуже часто не можна при цьому просто вводити необхідний продукт в їжу, оскільки він розкладається в травному тракті. В результаті необхідні постійні ін'єкції. Інша проблема - розробка методики точного дозування препаратів з урахуванням біохімічних показників хворого, їх змін протягом хвороби.
Замісна терапія успішно застосовується при лікуванні наступних спадкових захворювань:
- дефекти синтезу гормонів щитоподібної залози - препарати йоду, тиреоїдину;
- андрогенітальний синдром - глюкокортикоїди;
- дисгаммаглобулінемія - гамма-глобулін, поліглобулін;
- гемофілія А - переливання донорської крові, антигемофільний глобулін;
- хвороба Паркінсона - L-ДОФА (L-3-4-дигідроксіфенілаланін), який є попередником медіатора дофаміна;
- мукополісахаридози, міопатії – переливання плазми або лейкоцитної суспензії здорових людей (введення ферментів, яких не вистачає).
2. Вітамінотерапія.
Полягає у введенні вітамінів у великих дозах - в десятки або навіть сотні разів більших за фізіологічні. Дещо схожа на замісну терапію, але не така специфічна. Дія високих доз вітамінів обумовлена тим, що більшість з них є компонентами ферментів. Отже, дефект певного ферменту може бути частково компенсований за рахунок більш активного використання тих його кількостей, що наявні в організмі.
Вітамін В1 застосовується в терапії мегалобластичної анемії, підгострої некротизуючої енцефалопатії;
В6 - гомоцистинурії, судомного синдрому, гіпохромної анемії;
А - фолікулярного кератозу;
Д - рахіту;
Е - акантоцитозу.
3. Застосування індукторів або інгібіторів метаболізму.
До індукторів метаболізму відносять речовини, введення яких в організм сприяє посиленню синтезу деяких ферментів, а отже і підвищує інтенсивність відповідних метаболічних процесів. Приклади індукторів: фенобарбітал, левоміцетин, естрогени. Інгібітори, тобто речовини, що пригнічують обмінні процеси, застосовують тоді, коли необхідно загальмувати певну небажану біохімічну реакцію. Цей метод лікування зараз тільки проходить процес становлення, але вже зараз має певні успіхи в лікуванні таких хвороб, як подагра (пригнічення синтезу сечової кислоти), гепатолентикулярна дегенерація (регуляція обміну міді).
4. Хірургічне лікування.
Застосовується при лікуванні спадкових хвороб, пов'язаних з вадами розвитку: розщеплення губи та піднебіння, полідактилія, синдактилія, природжений стеноз привратника, природжений вивих кульшового суглобу. За останні десятиріччя стало можливим ефективно виправляти деякі природжені аномалії серця та магістральних судин, пересаджувати нирки.
Наступний напрямок - видалення (повне або часткове) аномально працюючих залоз: аденом паращитоподібних залоз, частини щитоподібної залози, гонад при порушеннях статевої диференціації.
При спадкових порушеннях імунітету можливе пересадження тімусу ембріонів, кісткового мозку.
5. Дієтотерапія, регулювання вживання ліків.
Суть цієї групи методів - обмеження надходження в організм тих речовин, обмін яких порушений, з метою запобігання розвитку хвороби.
При багатьох спадкових дефектах метаболізму є єдиним патогенетичним методом лікування, а в деяких випадках - і засобом профілактики. Найбільш важлива дієтотерапія для тих хвороб, які починають розвиватись від народження, оскільки вона дозволяє запобігти суттєвим порушенням онтогенезу.
При застосуванні дієтотерапії важливо адаптувати дієту до потреб організму, що росте, проводити регулярний клінічний і біохімічний контроль. Наприклад, лікування фенілкетонурії повинно полягати в повному виключенні фенілаланіну з раціону, тобто в годуванні спеціальними білковими гідролізатами. Але фенілаланін - це незамінна кислота, отже для відносно нормального фізичного розвитку він повинен надходити в організм з їжею. Таким чином, головна проблема при лікуванні фенілкетонурії - не допустити як розвитку хвороби, так і порушення росту дитини.
Встановлено, що вміст фенілаланіну в їжі повинен становити не більше 21% вікової фізіологічної норми. сучасні харчові раціони для хворих на фенілкетонурію дозволяють дозувати надходження фенілаланіну в організм у відповідності з його концентрацією в крові за даними біохімічного аналізу.
Рання діагностика і вчасно розпочате лікування (з перших 2-3 місяців життя) забезпечують нормальний розвиток дитини. Якщо лікування почате з 3-12 міс., імовірність успіху становить 26%, з 1-3 років - 15%.
При лікуванні дефектів обміну вуглеводів дієтотерапія дає добрі результати: виключення певного цукру з раціону далеко не таке небезпечне, як виключення амінокислоти. Галактоземія, непереносимість фруктози, порушення всмоктування окремих моно- та дисахаридів успішно лікуються відповідною дієтою за однієї умови - своєчасного початку лікування.
4. Медико-генетичне консультування при генних хворобах
Визначити ризик народження хворої дитини в наступних випадках:
а) у здорових батьків народилась дитина з аутосомно-рецесивним захворюванням;
б) один з батьків має аутосомно-домінантну хворобу;
в) обидва батьки здорові, але батькова мати має аутосомно-рецесивну хворобу, частота якої в популяції складає 1:10 000;
г) обидва батьки здорові, але дідусь матері має Х-зчеплене рецесивне захворювання;
д) обидва батьки здорові, але батько матері має аутосомно-домінантну хворобу з пенетрантністю 40%;
е) у здорових батьків народилась дитина з аутосомно-домінантною хворобою, яка у родоводі не зустрічається.
При вирішенні задач спирайтесь на матеріали попереднього заняття - Таблицю 1 "Типи генетичних задач у медико-генетичному консультуванні". Спробуйте самостійно проаналізувати задачі, визначити шлях їх розв'язання. На відеоконференції обговоримо.